Core Project Data
In their integrated DNA form - referred to as a provirus - retrovirus genomes are flanked at either side by identical long terminal repeat (LTR) sequences, each of which is composed of distinct U3, R, and U5 regions.
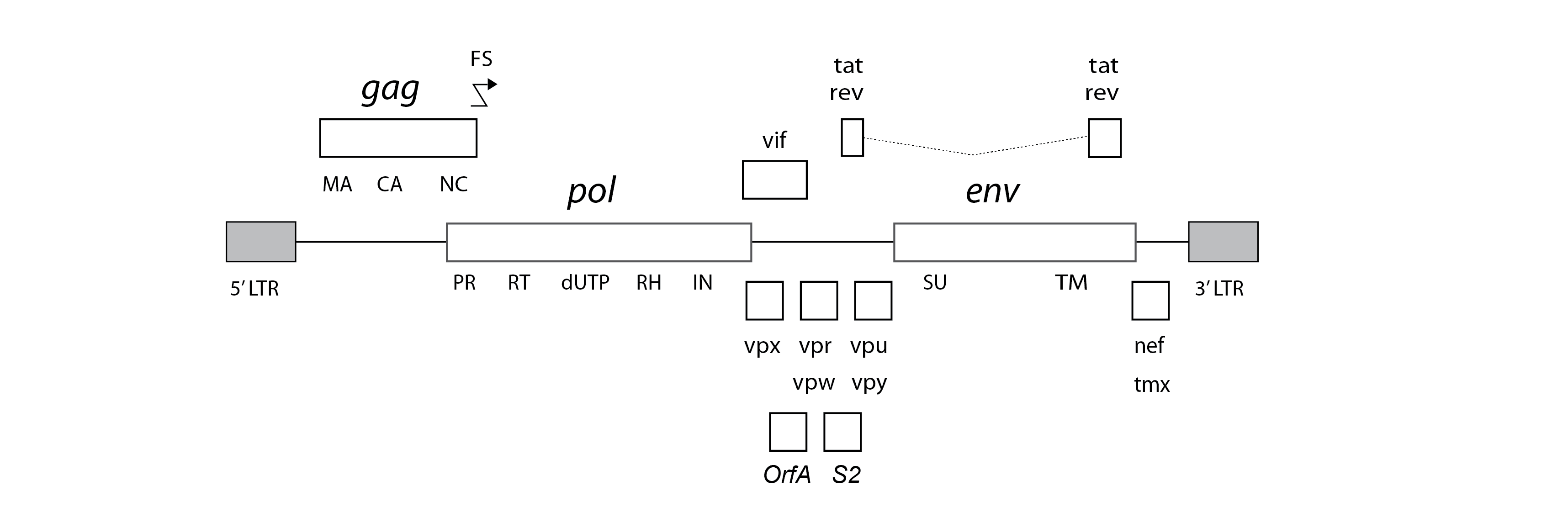
Lentiviruses typically encode a range of ‘accessory’ genes in addition to the fundamental gag, pol, and env genes encoded by all retroviruses. Among these, the rev gene is thought to be encoded by all lentiviruses, while tat is encoded by all except SRLVs and FIVs. Numerous other accessory genes have been defined, but the evolutionary relationships between these genes are not well-characterized.
A standard set of genome features for lentiviruses has been defined, reflecting current knowledge, and incorporated into Lentivirus-GLUE.
Lentivirus-GLUE contains master reference sequences for all known lentivirus species:
- Primate group: HIV-1 (AF033819)
- Equine group: EIAV (AF016316).
- Small Ruminant group: SRLV-A (NC_001452).
- Feline group: FIV (M25381).
- Bovine group: BIV (M32690).
Reference sequences are linked to metadata in tabular format.
We annotated the locations of genome features on master reference sequences.
Multiple sequence alignments (MSAs) are the basic currency of comparative genomic analysis. MSAs constructed in this study are linked together using GLUE's constrained MSA tree data structure.
A 'constrained MSA' is an alignment in which the coordinate space is defined by a selected reference sequence. Where alignment members contain insertions relative to the reference sequence, the inserted sequences are recorded and stored (i.e. sequence data is never deleted).
GLUE projects have the option of using a data structure called an alignment tree to link constrained MSAs representing different taxonomic levels, and we've used this approach in Lentivirus-GLUE.
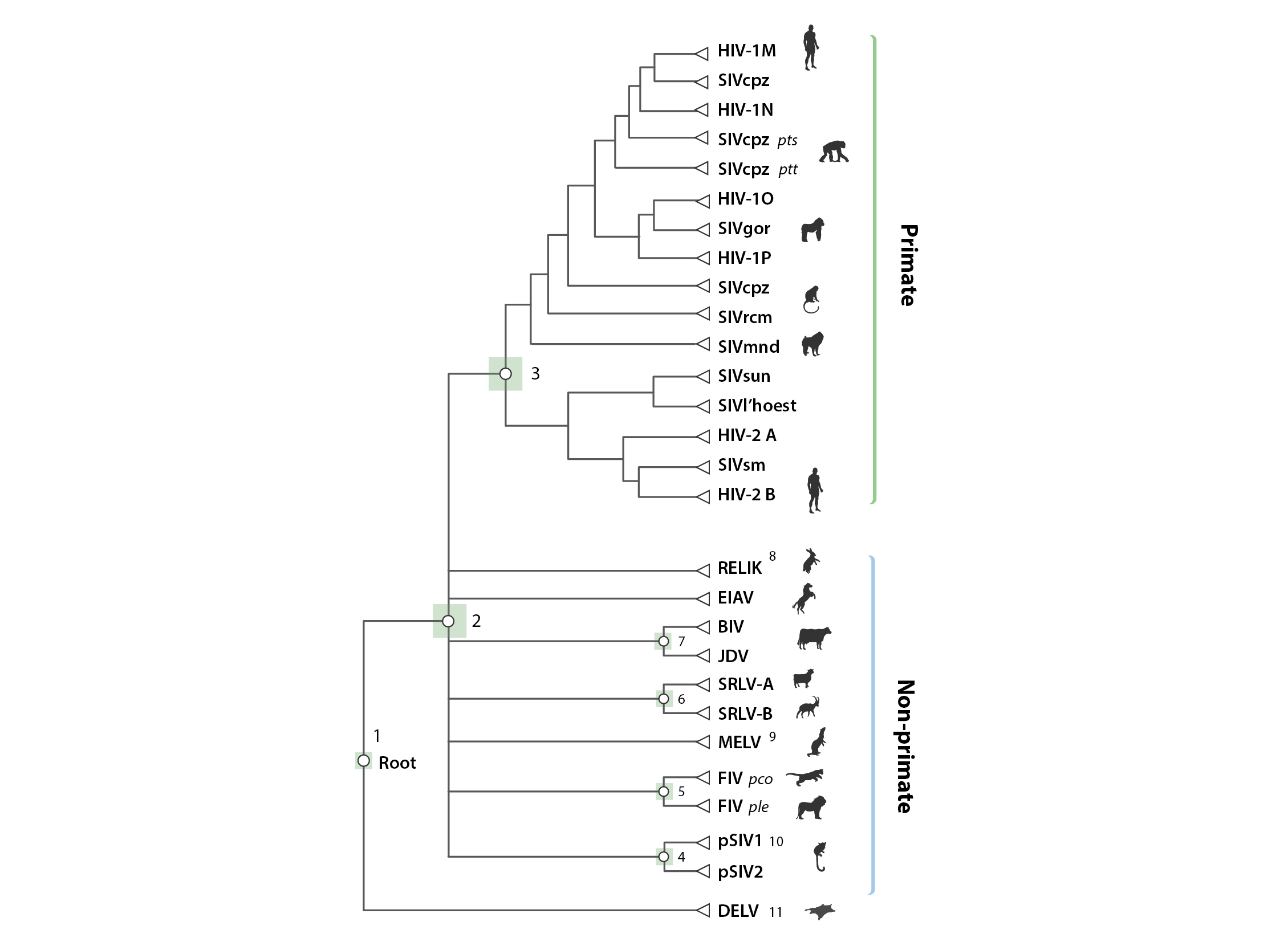
The schematic figure above shows the 'alignment tree' data structure currently implemented in Lentivirus-GLUE. We used an alignment tree data structure to link alignments, via a set of common reference sequences. The root alignment contains reference sequences for major clades, whereas all children of the root inherit at least one reference from their immediate parent. Thus, all alignments are linked to one another via our chosen set of master reference sequences.
Example alignments include:
- Root alignment: lentivirus-root-gagpol
- Genus-level alignments: Available here