-
Notifications
You must be signed in to change notification settings - Fork 0
/
Copy pathpart03.Rmd
257 lines (178 loc) · 11 KB
/
part03.Rmd
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
---
title: "Workflows, Pipeline and Workflow Managers"
author: "Mark Dunning - Sheffield Bioinformatics Core"
date: '`r format(Sys.time(), "Last modified: %d %b %Y")`'
output:
html_notebook:
toc: yes
toc_float: yes
css: stylesheets/styles.css
---
```{r setup, include=FALSE}
knitr::opts_chunk$set(echo = TRUE,eval=FALSE)
```

# Why do we need a pipeline? How might we create one?
We have now learnt a few commands that, when joined together, can form the basis for a minimal analysis pipeline
- use `fastqc` to create QC reports for each sample
- use `multiqc` to combine the QC
- index a reference transcriptome using `salmon`
- produce a set of quantified genes using `salmon`
The next natural step would be to record these commands in a *script* so that we can document our analysis and re-run as required. We don't usually have a text editor in our Unix environment, so it is good practice to create and edit the script using the command-line. The `nano` editor is commonly-found on Unix systems and running the command `nano` with a file name will create that file if it does not exist.
```{bash}
nano analysis_script.sh
```
**Enter the following in the nano window that appears**
```{bash}
fastqc fastq/*.fastq.gz
mkdir -p qc/
mv fastq/*.html qc/
mv fastq/*.zip qc
multiqc -f -o qc/ qc
```
Press **CTRL + X** following by **Y** (when asked if you want to save the modified buffer) to return to the terminal.
We can execute the script using the `bash` command
```{bash}
## remove qc directory first, so we can see the script in action
rm -r qc/
ls -l
bash analysis_script.sh
```
This will run through the QC steps of the pipeline. The next stage would be to add the steps for `salmon` quantification. Since we want the ability to re-run the whole pipeline from scratch if required, we need to add the step to create the salmon index
```{bash}
salmon index -i index/GRCh38_salmon -t ref_data/Homo_sapiens.GRCh38.cdna.chr22.fa
salmon quant -i index/GRCh38_salmon --libType A -r fastq/ERR732901_sub.fastq.gz -o quant/ERR732901
```
Our pipeline is already quite short and is running on a small dataset, but already it is taking a little while to run. The pipeline has been written in a linear fashion, so that each step must be completed in order. If our salmon alignment code needed to be changed we would have to re-run all the QC. This is not a huge problem here, but could be quite inefficient for a more-realistic dataset.
We also have a number of options for how to proceed with quantifying the remaining samples. The simplest approach copy-and-paste the `salmon quant` line with different sample names
```{bash}
salmon quant -i index/GRCh38_salmon --libType A -r fastq/ERR732902_sub.fastq.gz -o quant/ERR732902
salmon quant -i index/GRCh38_salmon --libType A -r fastq/ERR732903_sub.fastq.gz -o quant/ERR732903
###etc....
```
This is not particularly satisfactory as it is prone to typo errors. An alternative might be to employ a *for loop*, which you might have [come across previously](https://datacarpentry.org/shell-genomics/04-redirection/index.html). Ultimately, we will not do either as we are going to use a specific type of program for running our pipeline.
# Why do we need a workflow manager
As we have discussed, there are a number of options to extend our pipeline to multiple samples. These require more programming knowledge than we might be comfortable with. There are a few other issues with the script that we have created.
- As pipeline steps have to be re-run in sequence; even if the initial pipeline steps ran sucessfully they will still be re-run every time
- There is no error-checking.
- The pipeline will not neccesarily run on another environment as it will assume that the `fastqc`, `multiqc` and `salmon` tools can be found.
None of these issues are impossible to solve, but this isn't intended to be a workshop on software development best-practice.
**In reality, we would recommend people re-using existing analysis pipelines rather than writing their own**. We will look at an example using the nextflow workflow manager, although similar tools such as snakemake are also popular in Bioinformatics
- [nextflow](https://www.nextflow.io/)
- [snakemake](https://snakemake.readthedocs.io/en/stable/)
<div class="information">
nextflow is available as a *module* on the University of Sheffield Stanage HPC.
```
module load Nextflow/23.10.0
```
- [https://docs.hpc.shef.ac.uk/en/latest/stanage/software/apps/nextflow.html](https://docs.hpc.shef.ac.uk/en/latest/stanage/software/apps/nextflow.html)
- [Sheffield Bioinformatics Core documentation on nextflow and nf.core](https://sbc.shef.ac.uk/nf-core-user-docs/)
</div>
## Running a nf.core pipeline
In our opinion, nextflow is particular appealing as many popular Bioinformatics pipelines have already been written using nextflow and have been distributed as part of the nf.core project
- [nf.core homepage](https://nf-co.re/)
We will be showing the RNA-seq pipeline in particular
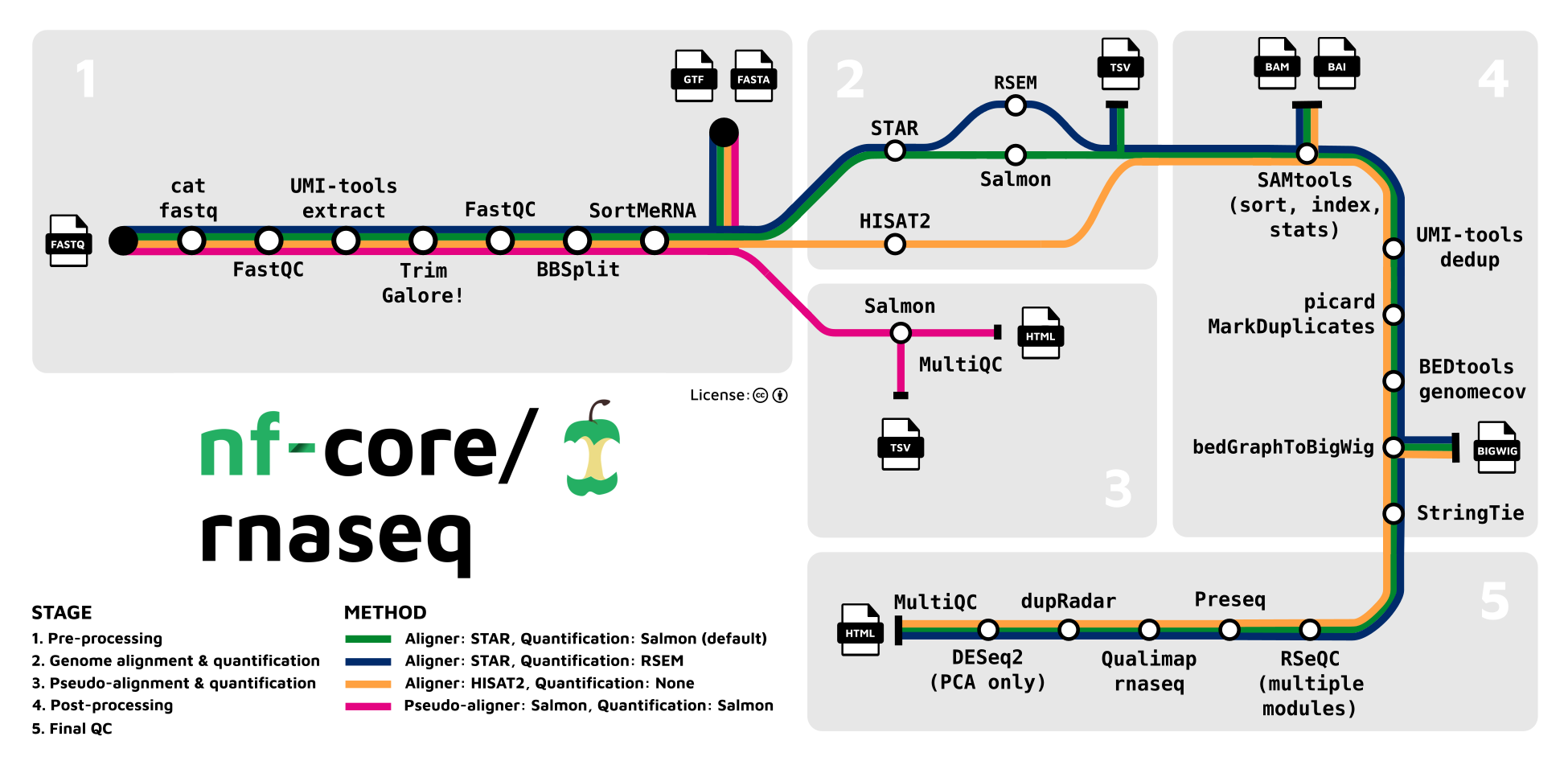
- [nf.core RNA-seq pipeline](https://nf-co.re/rnaseq)
A minimal number of options required to run an nf.core pipeline such as RNA-seq are:-
```{bash}
nextflow run nf-core/rnaseq \
--input samplesheet.csv \
--outdir <OUTDIR> \
--genome GRCh37 \
-profile docker\
```
where:-
- `nf-core/rnaseq` is a reference to the pipeline that we want to run
- `--input` is the location of a samplesheet defining the raw data to be processed
- `--outdir` is a directory that will contain the final results of the pipeline
- `genome` is the shorthand name for the genome to be used as a reference
- `profile` defines how software included in the pipeline is to be downloaded/installed (**See later**)
We have customised some of the options of the pipeline so run a reduced number of steps are run for the workshop, and using a custom genome containing a single chromosome.
```{bash}
cat run_nextflow.sh
```
The particular steps that we have modified are as follows:-
```
##use a particular pipeline version
-r 3.8.1 \
## Skip aligning to the whole genome
--skip_alignment \
## Skip trimming read sequences
--skip_trimming \
## use the salmon quantification tool
--pseudo_aligner salmon \
## Use our own set of references rather than downloading
--fasta ref_data/Homo_sapiens.GRCh38.dna_rm.chromosome.22.fa \
--transcript_fasta ref_data/Homo_sapiens.GRCh38.cdna.chr22.fa \
--gtf ref_data/Homo_sapiens.GRCh38.108.chr22.gtf \
--salmon_index index/GRCh38_salmon \
## restrict the amount of memory requested \
--max_memory 2GB
```
The files that we want to analyse are defined in a sample sheet. The format of the sheet is checked by the pipeline as one of the first steps. The column names have to match *exactly* what the pipeline expects.
```{bash}
cat nf_samplesheet.csv
```
We can run the pipeline as follows
```{bash}
bash run_nextflow.sh
```
The output from the workflow will be written to a directory `nf_results`, which doesn't need to exist before the pipeline has been run. You should also see that a `work` directory is created.
## Exercise
<div class="exercise">
**Exercise**
Run the script `run_nextflow.sh` (around ~5 to 10 minutes) and afterwards look at the output in the `nf_results` folder and familiarise yourself with the outputs. What extra steps have been performed in addition to the script that we created earlier?
</div>
## What software does nextflow use in it's pipeline?
You should notice that the nf.core pipeline has produced quantification files for each sample, and also combined the `salmon` outputs into a single file. It also run some QC plots from the DESeq2 R package. However, R is not part of our software environment. We can see this by running the following commands which would usually report the path that R is located at, or run the command-line R.
```
which R
R
```
nextflow has it's own way of installing and running software which does not depend on the operating system that is being used to run the pipeline. This is specified by this part of the run script.
- [nf.core profile options](https://nf-co.re/rnaseq/3.9/usage#profile)
```
-profile singularity
```
In fact, we could have run the pipeline if we didn't have `salmon` and `fastqc` installed. The implication of this being that you can re-run the pipeline on your own machine or HPC environment with minimal software installation. The only dependancies are `nextflow` itself (which in turn requires `java`) and some *containerisation* or package management software such as `singularity`, `docker` or `conda`. This is a significantly easier requirement to fulfill than having to install each piece of software individually.
<div class="information">
If you are using a HPC environment you will probably want to keep the singularity option in the profile.
</div>
## Re-running the pipeline
<div class="exercise">
**Exercise**
There are two fastq files in our `fastq` folder that have not yet been analysed; `ERR732908` and `ERR732909`. Use the `nano` editor to modify the samplesheet `nf_samplesheet.csv` to include `ERR732908`, and re-run the pipeline. What do you notice about the time taken to run the pipeline?
</div>
The pipeline should be quicker this time around. This is because we had all the software downloaded and installed from the previous run. However, it still re-analysed the first seven samples from the samplesheet - which is not ideal. If we want a report on what analyses have been performed and how long they took we can look at a report from the command-line
```
## You will have to use auto-complete and choose the most-recent report
cat nf_results/pipeline/execution_trace
```
There is also a HTML version of the report that we can view through the file system.
## Resuming a pipeline
Ideally, we would like the pipeline to detect what jobs have been run successfully and not repeat those jobs. The option `-resume` in nextflow will allow you to do this. (note the single `-` when adding this option). Modify the nextflow script with `nano` to contain the following lines at the top
```
nextflow run nf-core/rnaseq -profile singularity \
-resume \
-r 3.8.1 \
....
```
We can now edit the samplesheet again to process the sample `ERR732909`
```
sample,fastq_1,fastq_2,strandedness
ERR732901,fastq/ERR732901_sub.fastq.gz,,unstranded
ERR732902,fastq/ERR732902_sub.fastq.gz,,unstranded
ERR732903,fastq/ERR732903_sub.fastq.gz,,unstranded
ERR732904,fastq/ERR732904_sub.fastq.gz,,unstranded
ERR732905,fastq/ERR732905_sub.fastq.gz,,unstranded
ERR732906,fastq/ERR732906_sub.fastq.gz,,unstranded
ERR732907,fastq/ERR732907_sub.fastq.gz,,unstranded
ERR732908,fastq/ERR732908_sub.fastq.gz,,unstranded
ERR732909,fastq/ERR732909_sub.fastq.gz,,unstranded
```
and re-run the pipeline.
```
bash run_nextflow.sh
```
The pipeline now runs significantly quicker because it detects that all the analyses have been completed and *cached*. If we check the `execution_trace` text file for this latest run it should say that most tasks were `CACHED` - meaning that the results from the previous analysis was used.